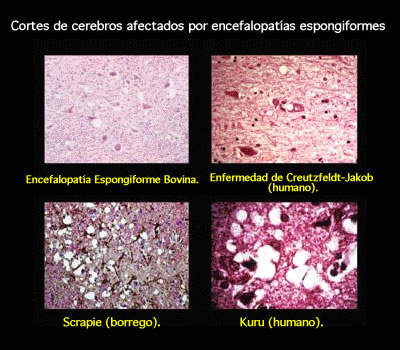
La causa son los priones, que son proteínas modificadas,infecciosas y malignas. La proteína prión se encuentra de forma natural en los mamíferos. La encefalopatía espongiforme aparece cuando la estructura normal de esta proteína prión se altera. La proteína prión alterada (PrPsc) actúa sobre las proteínas prión normales (PrP) y consigue que éstas cambien también su estructura. Poco a poco, aparecen malformaciones en las células del cerebro (neuronas). El prión alterado provoca "agujeros" en las neuronas. Visto al microscopio, el tejido cerebral adquiere la apariencia de una esponja. Y esta es la característica que da nombre a las encefalopatías espongiformes.

Son muy resistentes no pudiéndose eliminar, no hay tratamiento. Son resistentes a temperaturas superiores a 80ºC, a las radiaciones ultravioleta, al formaldehido (desinfectante), y no da lugar a ningún tipo de respuesta inmunitaria o inflamatoria y no se produce interferon (proteína producida por las células inmunitarias para defender al organismo de las infecciones).
Son muy resistentes no pudiéndose eliminar, no hay tratamiento. Son resistentes a temperaturas superiores a 80ºC, a las radiaciones ultravioleta, al formaldehido (desinfectante), y no da lugar a ningún tipo de respuesta inmunitaria o inflamatoria y no se produce interferon (proteína producida por las células inmunitarias para defender al organismo de las infecciones).

Existen tres variedades de enfermedad: Esporádica (sin factores de riesgo previos, es la mas corriente), hereditaria o familiar (el prion pasa de padres a hijos) y la adquirida (por exposición a tejido cerebral o del sistema nervioso infectado).
La enfermedad de Creutzfeld-Jakob forma parte del conjunto de encefalopatías espongiformes transmisibles o EET, entre las que también podemos encontrar la enfermedad de kuru, el insomnio familiar letal y la enfermedad de Gerstmann-Straussles-Sheinker (GSS).
El kuru se encontró en una tribu de Nueva Guinea, donde las mujeres y los niños comían los órganos de los hombres muertos y sufrían espasmos, alteraciones neurológicas y un largo etcétera. El insomnio familiar letal y la GSS son muy raras y hereditarias.
La variedad de encefalopatía espongiforme bovina o enfermedad de las vacas locas se encontró en Inglaterra, donde se vio que ponían trozos de ovejas muertas en los piensos de las vacas, y así pasaban los priones de un animal a otro y finalmente al hombre.
Realmente, todos poseemos priones, pero son proteínas normales que no causan daño, esto solo sucede cuando la proteína es modificada. En este tipo de enfermedades cuando entra un prion al organismo produce una especie de cascada exponencial. Es decir, un prion infecta a otro, y este va infectando a los demás y así sucesivamente.
El diagnostico de la enfermedad se puede hacer por biología molecular y, como he dicho anteriormente, no existe tratamiento. Lo mejor que se puede hacer es reducir riesgos realizando adecuadamente la desinfección del material quirúrgico en las operaciones.
No hay comentarios:
Publicar un comentario